News Detail
 来源:2026-05-11 11:23:18
来源:2026-05-11 11:23:18
 浏览量:19497
浏览量:19497
很多人以为,吃了药或者打了针,药物就会直奔病灶去「干活」。实际上,药物进入人体后,要走一条相当复杂的路:先被吸收进血液,再被运送到全身各处,接着在肝脏被「改造」变样,最后通过尿液或粪便离开身体。这条路上的每一个环节,都决定了药物能不能安全有效地发挥作用。
药代动力学(PK)研究的就是这个过程——药物在体内的「旅行日志」。而药物代谢动力学(DMPK)则更进一步,不仅记录旅行过程,还要弄清楚药物在旅途中「变身」成了什么,这些变身后的产物有没有危险。
爱思益普的 DMPK 平台,就是专门帮制药企业解答这些问题的。从早期筛选成百上千的化合物,到最终确定哪个分子能进入人体试验,DMPK 数据贯穿始终,相当于新药研发里的「导航仪」。
在药物研发初期,化学家们可能合成了几百个结构相似的新分子。这时候最迫切的问题是:哪些分子有“成药”的潜力?哪些连第一步都迈不出去?
爱思益普设计了一套“快速体检”方案,叫做 Tier 1 ADME 面板,专门给化合物做基础画像:
🔑 溶解度:药物能不能在水里化开?化不开的话,吃进去也吸收不了。
🔑 渗透性:用 Caco-2 肠道细胞模型模拟,看药物能不能穿过肠壁进入血液。
🔑 蛋白结合率:药物进入血液后,有多少被血浆蛋白“抓”住?抓得太紧,游离的药物就少,药效可能打折扣。
🔑 代谢稳定性:把药物放到人肝微粒体里“孵”一会儿,看它被代谢酶分解得多快。分解太快,药效持续时间短;分解太慢,可能蓄积中毒。
🔑 脂溶性:用 Log D 值衡量药物是“亲油”还是“亲水”,这直接影响它能穿过哪些生物膜。
这套「体检」用 LC-MS/MS(液相色谱-串联质谱)来检测,灵敏度高、速度快,能在很短时间内筛掉明显不合格的分子 。
通过初筛的化合物,再进入 Tier 2“深度体检”,重点查两件事:
📚 第一,药物相互作用风险。很多药物靠肝脏里的 CYP450 酶家族来代谢。如果一个新药抑制了 CYP3A4,那么患者同时服用经 CYP3A4 代谢的其他药物(比如某些降压药、抗生素)时,就可能因为代谢受阻而蓄积中毒。爱思益普通过 CYP 抑制实验和诱导实验,提前预警这类“撞车”风险 。
📚 第二,转运体影响。肠道和肝脏细胞膜上布满了各种转运蛋白,像 P-gp 和 BCRP 这类“外排泵”,会把药物“赶”回肠道或血液里,降低药效。了解化合物是否是这些泵的“目标”,对预测口服吸收和药物相互作用都很重要。
体外实验高效,但终究是在培养皿里模拟。药物在活的动物体内会怎么表现?这才是关键问题。爱思益普的体内 PK 平台支持小鼠、大鼠、犬、猴等常用实验动物 ,给药方式也很灵活:静脉注射(直接进血液)、口服(模拟人吃药)、皮下注射、腹腔注射、肌肉注射等 。根据研究目的,可以设计单次给药看“一次性”表现,也可以多次给药看长期蓄积情况。还有一种叫“盒式给药”的聪明办法——把多个化合物同时给一只动物吃,快速比较它们的 PK 特征,省时省力 。
动物实验的核心产出是一张“血药浓度-时间曲线”:
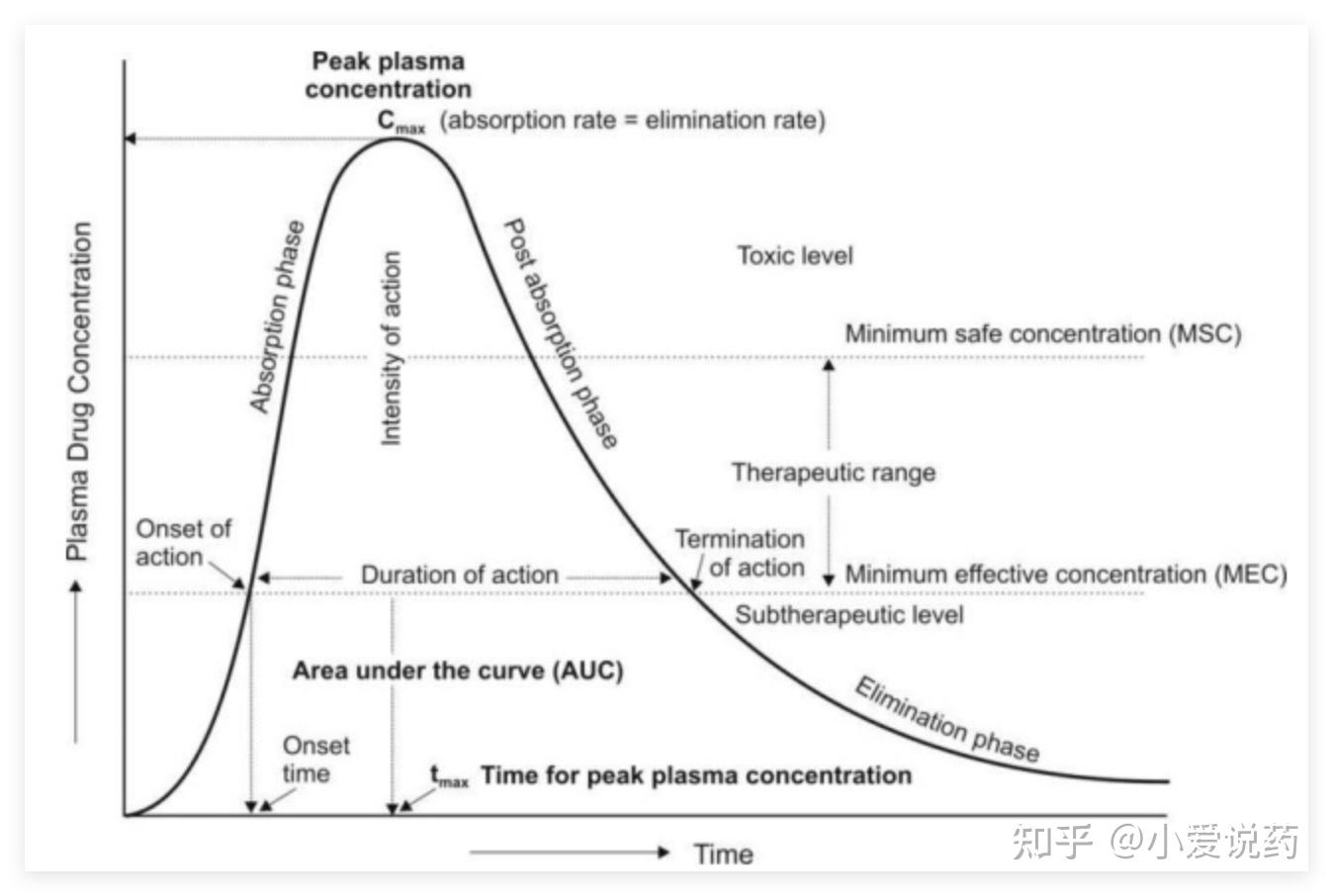
这张图详细标注了曲线的关键参数:Cmax是峰值浓度,tmax是达峰时间,AUC是曲线下面积(反映药物总暴露量),MEC是最低有效浓度,MSC是最低安全浓度。药物浓度必须维持在"治疗窗"内——太低没效果,太高有毒性。
从这条曲线可以算出几个关键数字:
🖥️ 半衰期(t1/2):药物浓度下降一半需要多久。这直接决定一天吃几次药。
🖥️ 清除率:身体「处理」药物的速度。清除太快,药效短;清除太慢,容易蓄积。
🖥️ 生物利用度:口服后真正进入血液的药量比例。很多药口服生物利用度不到 50%,意味着吃进去一半都浪费了。
🖥️ 分布容积:药物在体内是「集中」还是「分散」。分布广泛的药物可能更容易进入组织。
除了抽血测浓度,爱思益普还能做组织分布研究——把动物安乐死后,取心、肝、脾、肺、肾、脑、小肠、胃、脂肪、皮肤、睾丸、子宫等十余种组织,检测药物在各处的分布。这能回答两个重要问题:药物有没有在毒性靶器官(比如肝脏)蓄积?能不能到达需要治疗的组织(比如脑组织)?
药物进入体内后,很少以“本来面目”离开。肝脏里的代谢酶会把药物分子“改造”成各种代谢产物。有些变身后的产物活性更强,有些则带有毒性。所以,追踪药物的“变身轨迹”至关重要。
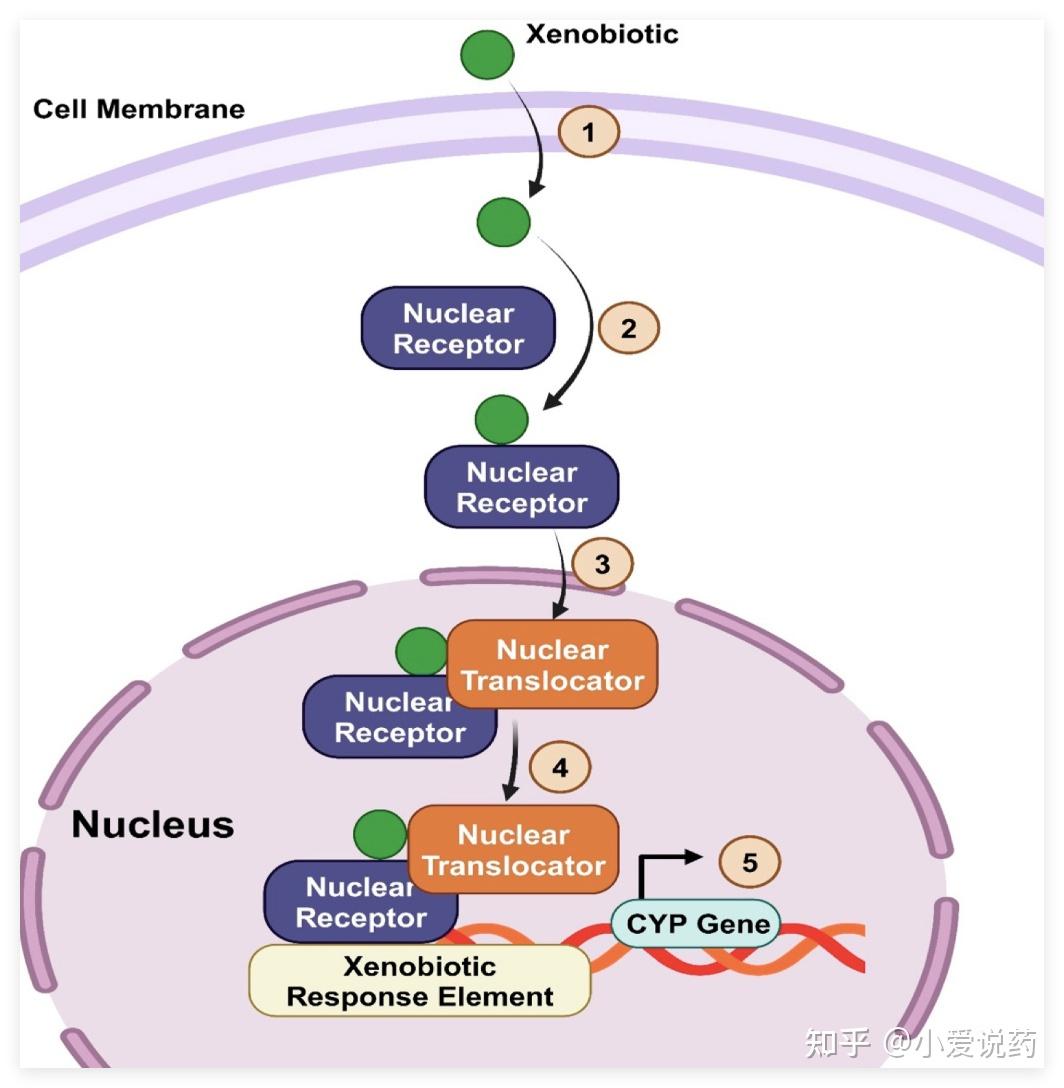
这张图展示了药物(绿色圆球)进入肝细胞后,如何通过核受体激活CYP450酶基因,启动代谢过程的分子机制。爱思益普的代谢产物鉴定(Met ID)团队配备了 Thermo Q Exactive Plus 高分辨质谱仪,这台仪器能精确到小数点后四位的分子量,哪怕代谢产物浓度只有 nM 级别(十亿分之一摩尔)也能「抓」出来 。配合 UHPLC 超高压液相色谱和 Compound Discoverer 智能分析软件,可以从复杂的生物样品(血浆、尿液、胆汁、肝微粒体孵育液等)中识别出代谢产物的结构。
爱思益普的代谢产物鉴定:
🚀 早期阶段:找到代谢“软点”——药物分子上最容易被代谢酶攻击的位置。化学家可以针对这些位置修改结构,让药物更耐代谢、作用更持久。
🚀 IND 申报阶段:监管机构要求证明实验动物的代谢方式与人相似。如果小鼠代谢某药物的方式和人完全不同,那么在小鼠身上做的安全性实验就无法预测人体反应。爱思益普通过体外跨种属代谢产物比较,帮客户选择最合适的动物种属。
🚀 安全预警:有些代谢产物虽然量少,但反应性极强,会与蛋白质或 DNA 结合形成“加合物”,可能引发肝毒性或致癌风险。通过谷胱甘肽(GSH)捕获实验,可以在代谢过程中“拦截”这些危险分子,提前亮红灯 。
近年来,靶向蛋白降解技术(PROTAC)成为新药研发的热门方向。这类分子像「双头胶水」——一头抓住致病蛋白,另一头抓住细胞的「垃圾回收系统」(E3 泛素连接酶),把致病蛋白标记为「垃圾」后降解掉。
但 PROTAC 分子在 DMPK 层面面临独特挑战:
💧 分子量大(通常 700 - 1000 道尔顿),远超传统小分子药物(通常<500),导致溶解度普遍偏低;
💧 结构复杂,体外渗透性数据很难预测体内真实情况;
💧 血浆蛋白结合率极高,游离药物浓度极低,给检测带来困难;
💧 代谢途径复杂,传统评价方法往往不够用。

临床前研究需要在多种动物种属中验证药物的安全性和有效性,包括小鼠、大鼠、兔、犬、猴等,不同种属的代谢特征存在差异。
爱思益普针对 PROTAC 的特性,建立了从体外 ADMET 到体内 PK/PD 的整合评价方案:
💡 体外层面,除了常规检测,还特别关注血浆蛋白结合率的准确测定(用平衡透析和超速离心两种方法交叉验证),以及转运体介导的药物相互作用风险。
💡 体内层面,通过整合药代动力学与药效动力学研究,评估药物在体内的实际表现。
更重要的是,爱思益普把 DMPK 与自身的靶点筛选平台深度打通。从蛋白制备、酶学方法开发、三元复合物形成、蛋白降解评价,到体外 ADME、体内 PK 及体内药理,形成了一站式服务 。客户不需要在多个供应商之间来回协调,数据连贯性更好,研发效率更高。
DMPK 研究最终要回答的,不是「药物在体内发生了什么」,而是「基于这些数据,我们该怎么决策」。爱思益普的 DMPK 服务强调「系统化的数据解读」。
专业团队不仅提供原始数据,还会结合项目背景给出针对性建议 :
❓️ 如果一个化合物口服生物利用度只有 5%,是放弃还是改成注射给药?
❓️ 如果药物在肝脏浓度是血浆的 20 倍,是警示肝毒性风险,还是庆幸找到了治疗肝病的候选分子?
❓️ 如果代谢产物鉴定发现人特有而动物没有的代谢途径,该如何调整申报策略?
在合规层面,平台的服务设计遵循 NMPA(中国药监局)和 FDA 的指导原则。SLC 转运体研究符合 FDA 推荐规范 ,体内 PK 数据包满足 IND 申报要求 ,Met ID 提供动物种属选择的科学依据。对于需要放射性标记(如¹⁴C)的物料平衡研究,平台也具备相应能力。
合作模式上,爱思益普提供两种选择:
☂️ FTE 模式像“雇佣一个专属团队”,适合长期深度合作;
☂️ FFS 模式像“按单点菜”,适合单次或阶段性需求。无论 Biotech 初创公司还是大型药企,都能找到适合的合作方式。
新药研发的成功率不到 10%。DMPK 的价值,在于尽可能早、尽可能准地识别那 90% 的“失败者”,让有限的资源聚焦于真正有望成药的分子。这不是简单的“筛掉坏的”,而是用数据为每一个决策提供科学依据——从化学家该修改哪个结构,到项目经理该推进哪个分子,再到注册部门该选哪种动物做安全性实验。
爱思益普的 DMPK 平台,通过体外快速筛选、体内深度验证、代谢产物精细追踪,以及与特殊分子类型和跨平台服务的有机整合,构建了一条从实验室到申报资料的「数据高速公路」。在这条路上,每一个实验结果都是导航信号,每一组数据都在为最终的临床成功增加概率!

业务咨询
北京
业务咨询专线:010-6780-9840
联系地址:北京市经济技术开发区科创十三街18号院锋创科技园16号楼
上海
业务咨询专线:010-6780-9840
联系地址:上海市浦东新区蔡伦路780号新技术推广大楼3E5O室
徐州
业务咨询专线:010-6780-9840
联系地址:江苏省徐州市云龙区淮海文博园 二号楼2层
贵州
业务咨询专线:010-6780-9840
联系地址:贵州省贵阳市南明区龙岭路50号 欧美医药产业园一期2号楼